PROTEUS2 Help
PROTEUS2 is a web server that allows prediction of secondary structure and tertiary structure with a high degree of accuracy, without the user having any prior knowledge of the structure of the protein.
Video tutorial
How to use PROTEUS2
PROTEUS2 takes as input a FASTA file of single or multiple sequences or raw sequence data, and you can either upload a file that is stored on your computer containing the data, or paste the file directly into the entry box.Email options
You may also specify if you want the results emailed to you, or you can choose to watch a live progress report of your prediction. Due to the length of time that the prediction requires, it is recommended that you use the email option. Please note that if you do choose to use the email option, you will receive a link to a results page, not the results themselves.Method options
PROTEUS2 performs signal peptide prediction using PSignal, a locally developed tool. Users must select which source organism (gram-, gram+, euk) before submitting their sequences for analysis. PROTEUS2 uses a default of gram-. Users also have the option of running only secondary structure prediction or homology modelling or both. The default option are secondary structure prediction and homology modelling. Users also have the homology modelling energy minimization option. The default is turned on. Normally PROTEUS2 automatically selects PDB homolog for homology modelling. However users do have the option for overwriting PROTEUS2 and select their own template file. This may be done by entering a PDB accession number or selecting a properly formatted PDB file uploading it using browser button.Output
PROTEUS2 will output the following items:- Complete prediction information
- Graphical alignment of soluble PDB homologs
- Graphical alignment of transmembrane PDB homologs
- Legend information
- Signal peptide prediction
- Transmembrane prediction
- 3D-to-2D mapping for top-scoring transmembrane homolog
- Secondary structure prediction
- Detailed secondary structure prediction from each method used (PSIPRED, JNET, TRANSSEC)
- 3D-to-2D mapping for top-scoring homolog
- PSI-BLAST results
- Tertiary structure prediction, with coordinates. You can download the resulting PDB file, view the file in the browser, or view the image via WebMol (Java required).
H = Helix
E = Beta Strand
C = Coil
T = Membrane helix
B = Membrane strand
S = Signal peptide
c = Cleavage site
How PROTEUS2 measures up
Evaluation Details
|
||
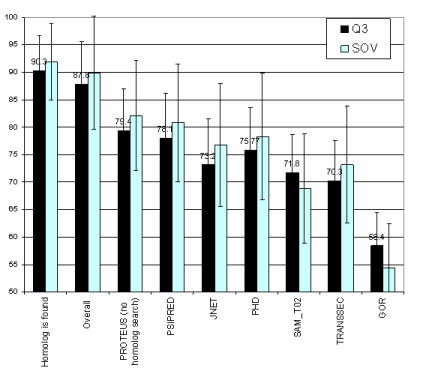
| Histogram comparing the Q3 (black) and SOV (gray) scores of PROTEUS2 (left 3 bars) versus PSIPRED, JNET, PHD, SAM-T02 and GOR. | ||
| ||
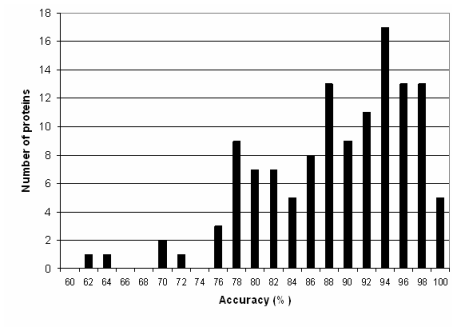
| Bar graph showing the performance of PROTEUS2 in predicting the secondary structure (Q3 score) for 125 proteins that were in neither the training or testing sets. |
How PROTEUS2 works
PROTEUS2 is comprised of seven separate processes that combine to produce the final output. See a flow chart below.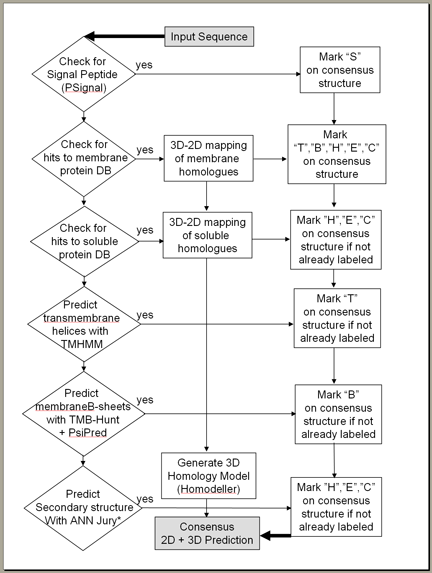
Methods
After the signal peptides and transmembrane segments (helices or beta-strands) have been identified, PROTEUS2 proceeds to the third structure prediction step. In this third step, PROTEUS2 initially compares the query sequence to a local, non-redundant version of the PDB containing nearly 13,000 sequences of water-soluble (i.e. non-membrane) proteins. The secondary structures for these proteins were assigned using VADAR. If a significant (Expect < 1e-10) match is found, the same 3D-to-2D mapping procedure is used to assign or predict the secondary structure. If no match can be found, the PROTEUS2 applies a locally developed "jury of experts" prediction method to predict the secondary structures as described in our earlier publication (Montgomerie et al., 2006).
Once the signal peptide, membrane spanning regions and secondary structures have been predicted, the query sequence is then directed to a locally developed homology modeling program called HOMODELLER. If a PDB file with >35% sequence identity was previously found in either of the two 3D-to-2D mapping steps, this structure is used as a template for HOMODELLER to build a 3D structure. While PROTEUS2 always succeeds in generating linear (i.e. secondary structure) predictions for any query sequence, 3D structures will only be generated if the query protein passes the HOMODELLER thresholds.
In developing the hidden Markov models for signal peptide prediction program (called PSignal or predictSP), more than 2000 signal peptides and 1200 control peptides (covering the N-terminal sequence to the cleavage site) were obtained from the SignalP data set. These examples were further partitioned into signal peptides belonging to Gram+, Gram- and Eukaryotic organisms. The predictor was trained to recognize not only the existence of a signal peptide, but also its length and cleavage site. PredictSP was subject to 10-fold cross validation during the testing phase.
The transmembrane prediction program builds from two previously developed and freely available programs, TMHMM and TMB-Hunt. TMHMM uses a hidden Markov model to identify transmembrane helices, while TMB-HUNT uses amino acid composition statistics to identify potential membrane beta-barrel proteins. We enhanced the performance of both methods by exploiting the 3D-to-2D mapping methods that we previously developed for predicting the secondary structure of water soluble proteins. To do so we created a database of 250 membrane proteins of known secondary and tertiary structure, with the membrane helices and membrane beta-strands appropriately labeled. As before, secondary structures that were derived from homologous proteins in the membrane protein database were given precedence over the predictions derived from TMHMM or TMB-HUNT. The program's performance was assessed using the TMH-Benchmark server (for transmembrane helices) and a 5X cross-validated subset of 40 transmembrane beta-barrels derived from the PDB.
In developing the "jury of experts" approach for predicting the secondary structure of soluble proteins we used the same programs and methods described previously (Montgomerie et al., 2006). The one change to this method involved editing the PDB-derived secondary structure database so that it contained only water-soluble proteins. This involved removing some 250 membrane proteins from the 13,000 protein data set. In training and testing the program nearly 2000 sequence-unique sequences were analyzed, requiring some 100 hours of CPU time. The program was written such that secondary structure predictions from the consensus predictor could be over-ridden if a homologous protein could be found in PROTEUS2's secondary structure database.
The homology modeling program, HOMODELLER, is quite conventional and employs standard homology modeling techniques. It uses the BLAST to search through a non-redundant version of the PDB database (which is updated weekly) to find and align the closest matching sequence homologue. Mismatched residues in the template sequence are changed to match the query sequence, but with the same x1 angles of the template. Gaps in coil regions are handled using a loop library (consisting of >13,000 loops derived from high resolution structures in the PDB). The inserted regions are superimposed and then iteratively adjusted to fit the surrounding regions using a simplex minimizer working in torsion angle space. The resulting structure is energy minimized using a torsion angle minimizer that uses cyclic coordinate descent in combination with a simple genetic algorithm. The method has been extensively tested and refined using hundreds of known structures. More recently, HOMODELLER was used to model more than 100,000 protein structures for the BacMap project.
Neural Network architecture
The methods and underlying theory to PSIPRED and JNET have been published previously and the programs were used as received without further modification. The TRANSSEC program was developed in-house using a Java-based neural network package known as Joone.TRANSSEC's underlying approach is relatively simple, consisting of a standard PSI-BLAST search integrated into a two-tiered neural network architecture. The first neural network operates only on the sequence, while the second operates on a 4 x N position-specific scoring matrix consisting of the secondary structure determined via the first network. The first neural net uses a window size of 19, and was trained on sequences from the PROTEUS2-2D database (independent from those used in training the other neural nets). This neural net had a 399-160-20-4 architecture (21 x 19 inputs, 2 hidden layers of 160 and 20, and four outputs). The second neural network used a position-specific scoring matrix, combining evolutionary information from a PSI-BLAST search, and structure information from the first neural network. It was also trained on a set of sequences from the non-redundant database mentioned above, and achieved a Q3 score of 70% and a SOV score of 72%. It used a window size of 9, and was based on a 36-44-4 architecture.
PROTEUS2's globular protein secondary structure method uses neural network mehtod that combines Psipred, JNet, and TRANSEC. The Jury-of-experts program, which combined the results of the three stand-alone secondary structure predictions was also developed using Joone. It consisted of a standard feed-forward network containing a single hidden layer. Using a window size of 15, the structure annotations and confidence scores from each of the three methods (JNET, PSIPRED, and TRANSSEC) were used as input.
Neural Network Training
The neural net was trained on 100 sequences chosen randomly from the non-redundant database mentioned above. Four output nodes were used, one for each of helix, strand or coil, as well as a fourth denoting the beginning and end of the sequence. A back-propagation training procedure was applied to optimize the network weights. A momentum term of 0.2 and a learning rate of 0.3 were used, and a second test set of 20 proteins was applied at the end of each epoch, to ensure that the network was trained for the most optimal number of iterations.Merging the homologous sequence with a prediction
If a homologous sequence was found whose sequence did not cover the entire length of the query sequence, then it was necessary to merge the homologous structure with that of the predicted structure. Though some secondary structure is the result of global interactions (hydrogen bonding between different parts of the sequence), homologous regions likely have similar local structure. Given that a homologous structure is likely to be more correct than the predicted structure, the homologous structure is mapped directly onto the predicted structure, replacing the states of the predicted structure with the homologous structure.Scoring and Confidence Values
PROTEUS2 uses the de novo secondary structure prediction programs JNET (Cuff JA, Barton GJ. Application of multiple sequence alignment profiles to improve protein secondary structure prediction Proteins. 2000 Aug 15;40(3):502-11), PSIPRED (McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000 Apr;16(4):404-5), TRANSSEC (Montgomerie S, Sundararaj S, Gallin WJ, Wishart DS. Improving the accuracy of protein secondary structure prediction using structural alignment. BMC Bioinformatics. 2006 Jun 14;7:301) and TMHMM Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001 Jan 19;305(3):567-80). Each of these methods generates residue-specific confidence values. These values are dependent on a number of factors specific to the predictor's design, but they largely reflect the degree of local sequence conservation from the multiple sequence alignments, individual amino acid secondary structure propensities and multi-residue secondary structure propensities. These program-specific confidence values are normalized by PROTEUS2 to integer values between 0 (low confidence) and 9 (high confidence). Additionally, the secondary structures assigned by template-based mapping (or structure-based mapping) in PROTEUS2 are given residue-specific confidence values (ranging from 7 to 9) and during the merge phase of PROTEUS2 they will overwrite in the confidence values in regions for which a suitable homolog has been found. The global confidence score is generated by summing all the residue-specific (integer) values over the entire sequence and dividing by the length of the sequence, giving a percentage. Generally predicted structures with global confidence scores of 80% or better can be considered to be quite reliable.
The global confidence score is generated by summing all the residue-specific (integer) values over the entire sequence and dividing by the length of the sequence, giving a percentage. Generally predicted structures with global confidence scores of 80% or better can be considered to be quite reliable.
Scoring Definitions
Q3 = overall three-state per-residue accuracy
Q3 = (#correct α helix + #correct β strand + #correct coil)/length of protein
Q2 = overall two-state per-residue accuracy
Q2 = (#correct membrane + #correct non-membrane)/length of protein
SOV = overall three-state per-segment accuracy (see Zemla A, Venclovas C, Fidelis K, Rost B. A modified definition of Sov, a segment-based measure for protein secondary structure prediction assessment.
Proteins. 1999 Feb 1;34(2):220-3.)
SOV = 1/N Σ(i) Σ(Si) [(MINOV(S1:S2) + DELTA(S1;S2))/MAXOV(S1;S2)]*LEN
Where:
S1 and S2 are the observed and predicted secondary structure segments (in state i, which can be either H, E or C);
LEN(S1) is the number of residues in the segments S1;
MINOV(S1;S2) is the length of actual overlap of S1 and S2, i.e. the extent for which both segments have residues in state i, for example H;
MAXOV(S1;S2) is the length of the total extent for which either of the segments S1 or S2 has a residue in state i;
DELTA(S1;S2) is the integer value defined as being equal to the MIN{(MAXOV(S1;S2)- MINOV(S1;S2)); MINOV(S1;S2); INT(LEN(S1)/2); INT(LEN(S2)/2)}
Sensitivity = TP/(TP + FN) where TP is # of true positives and FN is # of false negatives
Specificity = TN/(TN + FP) where TN is # of true negatives and FP is # of false negatives
In information theory or computing science, Sensitivty = Recall
In information theory or computing science, Specficity = Precision
Energy Minimization and Energy Values
If a homology model is generated by PROTEUS2 and the energy minimization option is used (both are turned on by default), energy values of the structure (before and after minimization) are given as part of the output. PROTEUS2 employs a torsion-angle-based energy minimization that uses a genetic algorithm to sample conformation space (GAfolder). The method is similar to that employed by GENFOLD (Bayley MJ, Jones G, Willett P, Williamson MP. GENFOLD: a genetic algorithm for folding protein structures using NMR restraints. Protein Sci. 1998 Feb;7(2):491-9.). The potential energy function is a knowledge-based potential that includes information on predicted/known secondary structure, radius of gyration, hydrogen bond energies, number of hydrogen bonds, allowed backbone and side chain torsion angles, atom contact radii (bump checks), disulfide bonding information, and a modified threading energy based on data from Bryant and Lawrence (Bryant SH, Lawrence CE. An empirical energy function for threading protein sequence through the folding motif. Proteins. 1993 May;16(1):92-112.). The GAfolder energy function was trained and refined using ~35,000 3D decoys available through the PPT-DB website. Evaluations using nearly 27,000 3D structure predictions from the CASP7 collection showed that the potential was able to identify the native or near-native (<2.5 Angstroms RMSD) structure in 80/90 (89%) of the CASP7 targets. It was also able to identify native or near native structures 99.5% (26,800/26,900) of the time when challenged with 3D decoys. The energy values presented in PROTEUS2 correspond to average per-residue energies. They do not correspond to real energy units, although a negative value (< -1) is a good indication of a high quality structure.